

Gaucher Disease is an inherited metabolic disorder characterized by the buildup of harmful amounts of certain fats (lipids), specifically the glycolipid glucocerebroside, throughout the body, most notably in the bone marrow, spleen, and liver. It’s one of the most common Lysosomal Storage Disease in Malaysia and Singapore with 3 occurrences in every 1 000 000 births.
Symptoms
Gaucher disease symptoms and physical findings differ greatly between patients. Some people have few or no symptoms (asymptomatic), while others may have serious complications.
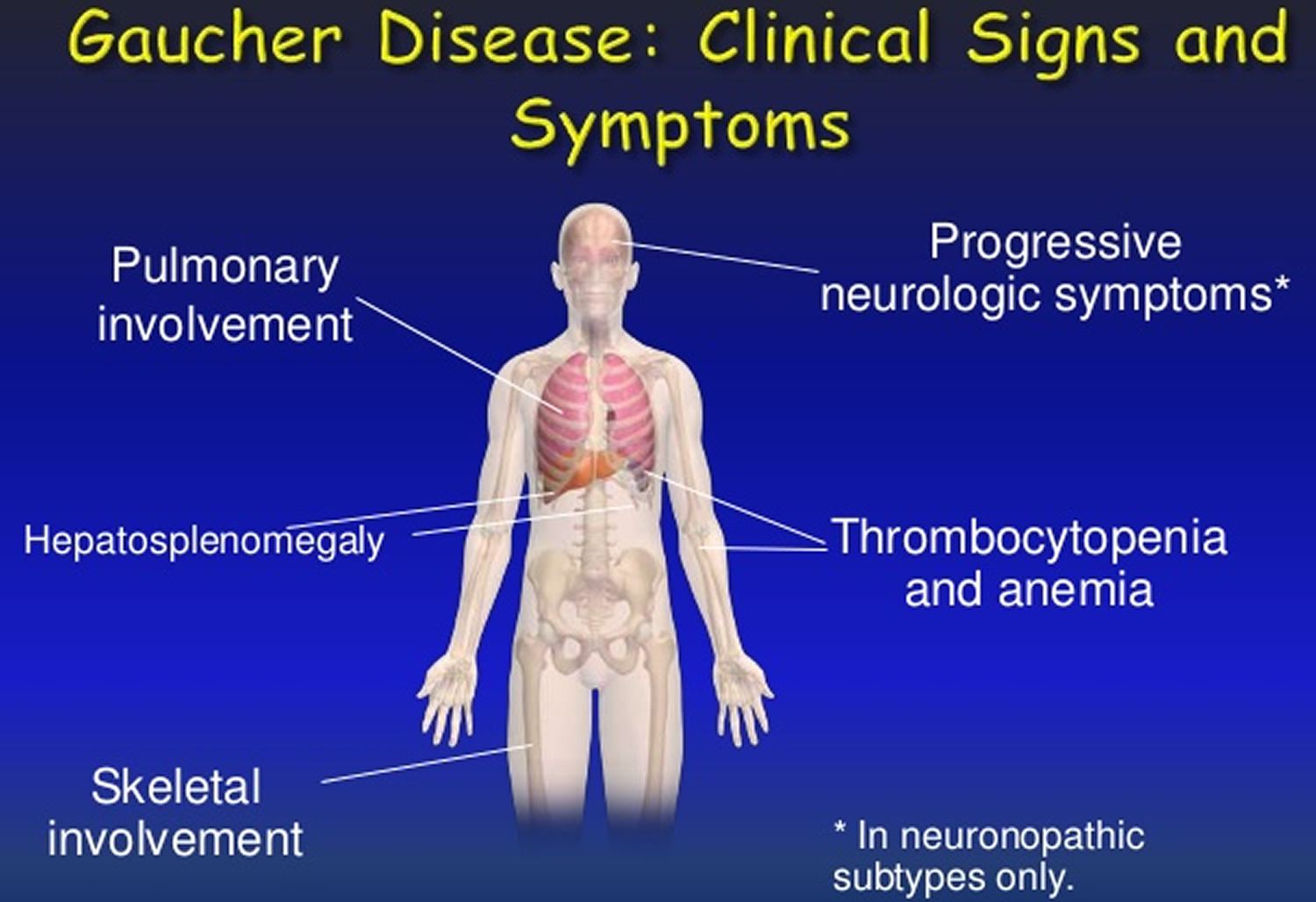
Common symptoms include an abnormally enlarged liver and/or spleen (hepatosplenomegaly), low levels of circulating red blood cells (anemia), low levels of platelets (thrombocytopenia), and skeletal abnormalities. Platelets are blood cells that help with clotting, and patients with thrombocytopenia may experience bleeding issues.
There are three types of Gaucher disease, each distinguished by the absence or presence and extent of neurological complications. Gaucher disease is inherited in three forms, all of which are autosomal recessive. Autosomal recessive is one of several ways that a trait, disorder, or disease can be passed down through families. This means two copies of an abnormal gene must be present in order for the disease or trait to develop.
Gaucher disease type 1 is the most common form of the disease in Western countries, accounting for approximately 95% of patients. Symptoms include enlarged spleen and liver, bone problems, and fatigue.
Gaucher disease type 2 is uncommon and is characterized by severe neurological (brain stem) abnormalities. It is typically fatal within the first two years and is currently incurable due to severe, irreversible brain damage.
Gaucher disease type 3 is the most common worldwide. It is intermediate in severity, causing symptoms similar to type 1 as well as some neurological involvement. While patients typically have a shorter lifespan, with treatment, some can live into their 50s.
Diagnosis
A blood test that measures glucocerebrosidase enzyme activity is currently used to diagnose Gaucher disease. For people with a known family history of Gaucher disease, a diagnosis can be confirmed with genetic testing. Individuals with a family history of the disease can also be tested for carriers. If both the child’s mother and father are carriers, the child has a 25% chance of being affected by Gaucher disease and a 50% chance of being a carrier. Prospective parents with a family history of Gaucher disease should seek genetic counseling, and prenatal testing can determine whether a fetus in the womb has Gaucher disease.
Treatment
Currently there is no cure for Gaucher Disease, however the disease’s symptoms can be treated to relieve the patient’s pain. Treatments include:
- Enzyme replacement therapy, which is effective for types 1 and 3
- Medicines
- Regular physical exams and bone density screening to check your disease
- Bone marrow transplant
- Surgery to remove all or part of your spleen
- Joint replacement surgery
- Blood transfusions
Witty Charman Medical shall be starting Diagnostic Testing services in newborn children and babies for this disease, using Dried Blood Stains in collaboration with College of American Pathology accredited Laboratories. Call to Wendy (+6012-387 8395), Auni (+60 13-233 3646) & Arissa (+60 12-210 3438) or write to wendy@wittycharman.com, info@wittycharman.com for further details.
References:
Hopkins Medicine. (2019, November 19). Gaucher Disease. Johns Hopkins Medicine. https://www.hopkinsmedicine.org/health/conditions-and-diseases/gaucher-disease
Mayo Clinic. (2022, April 30). Gaucher disease – Symptoms and causes. https://www.mayoclinic.org/diseases-conditions/gauchers-disease/symptoms-causes/syc-20355546
NORD – National Organization for Rare Disorders. (2020, March 27). Gaucher Disease. https://rarediseases.org/rare-diseases/gaucher-disease/