

Lysosomes are membrane-enclosed organelles that contain an array of enzymes capable of breaking down proteins, nucleic acids, carbohydrates, and lipids.
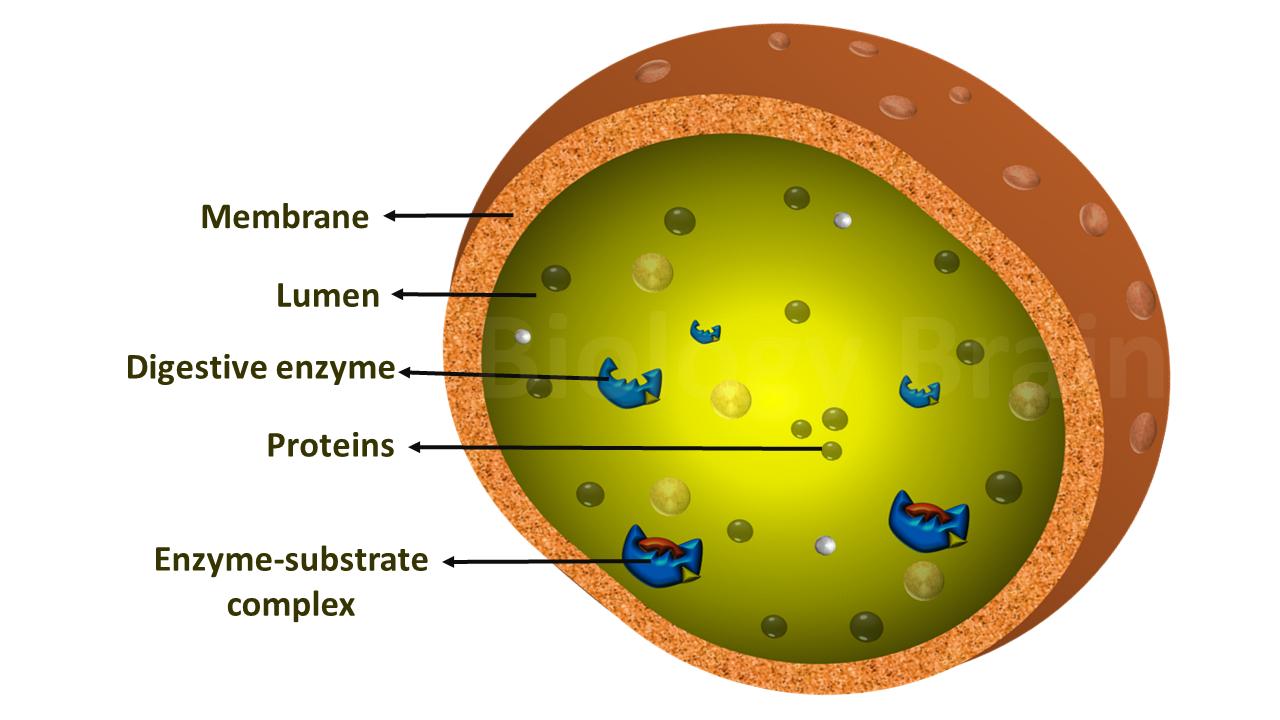
Lysosomes are enzyme sacs within cells that digest large molecules and recycle the fragments to other parts of the cell. Several critical enzymes are required for this process. If one of these enzymes becomes defective as a result of a mutation, the large molecules accumulate within the cell, eventually killing it.
Lysosomal storage diseases (LSD) are a group of inherited genetic disorders caused by defective lysosomal metabolism and subsequent substrate accumulation. The abnormal accumulation of various toxic materials in the body’s cells results from enzyme deficiencies. These include lipids, mucopolysaccharides (MPS), cholesterol ester, glycogen, and mucolipids.
There are nearly 50 of these disorders in total, and they can affect various parts of the body such as the skeleton, brain, skin, heart, and central nervous system. Up to this day, new lysosomal storage disorders are constantly being discovered.
Common types in Malaysia
In Malaysia, birth prevalence for all LSD is 0.93 per 100,000 live births with Gaucher disease being the most frequent LSD at 0.31 per 100,000. Metachromatic leukodystrophy has a prevalence of 0.2 per 100,000 births. In the mucopolysaccharidoses (MPS) group, all MPS birth prevalence total of 0.59 per 100,000 , where type IV has the highest calculated birth prevalence at 0.33 per 100,0001 live births followed by type II at 0.23 per 100,000
Gaucher Disease Type I,II and III

The most common type of lysosomal storage disorder is Gaucher disease. The majority of those affected are type I, and they may experience easy bruising, chronic fatigue, and an abnormally enlarged liver and/or spleen. Gaucher disease type II is characterized by neurological complications such as involuntary muscle spasms, difficulty swallowing, and the loss of previously acquired motor skills in newborns and infants. Gaucher disease type III manifests itself during the first ten years of life.
Metachromatic leukodystrophy

Early signs and symptoms of this disorder can be vague and gradual, making it difficult to diagnose. Walking unsteadiness is frequently the first symptom noticed. Occasionally, developmental delay or poor academic performance is the first symptom. Symptoms may include severe spasticity, seizures, and profound mental retardation over time.
Mucopolysaccharidoses Diseases
MPS diseases include Hurler Disease and variants, Hunter, Sanfilippo Types A, B, C, D, Morquio Types A and B, Maroteaux-Lamy, and Sly diseases. They are caused by disruptions in the normal breakdown of mucopolysaccharides, which are complex carbohydrates. They all share certain characteristics, such as bone and joint deformities that impair mobility and frequently cause osteoarthritis. Except for Sanfilippo disease, the diseases interfere with growth, resulting in short stature.
Diagnosis and Treatments
Prenatal diagnosis is possible for all lysosomal storage disorders. Early detection of lysosomal storage diseases, whether before birth or as soon as possible afterward, is important because when therapies are available, either for the disease itself or for associated symptoms, they may significantly limit the long-term course and impact of the disease.
While clinical trials on potential treatments for some of these diseases are currently underway, there is currently no approved treatment for many lysosomal storage diseases.
Witty Charman Medical shall be starting Diagnostic Testing services in newborn children and babies for this disease, using Dried Blood Stains in collaboration with College of American Pathology accredited Laboratories. Call to Wendy (+6012-387 8395), Auni (+60 13-233 3646) & Arissa (+60 12-210 3438) or write to wendy@wittycharman.com, info@wittycharman.com for further details.
Reference:
Omar, A., Shakrin, N. M., Aziz, N. A. A., Wahab, S. A. A., Hian, L. S., Yaakob, Y., & Jalil, J. A. (2018). Prevalence of Lysosomal Storage Diseases in Malaysia. Asian Journal of Medicine and Biomedicine, 38-38.
NORD – National Organization for Rare Disorders. (2017, August 28). Lysosomal Storage Disorders. https://rarediseases.org/rare-diseases/lysosomal-storage-disorders/