

Before we jump dive right in, a lot of you may wonder what is Mucopolysaccharidoses (MPS) Disease? Most if not, all might never even hear of this disease.
MPS are a group of disorders that fall under the inherited lysosomal storage disorders (LSD). LSD is caused by the defective function of lysosomes that is due to inborn errors which lead to the accumulation of excess substrates in cells of the organ. MPS was known to be inherited due to autosomal recessive traits.
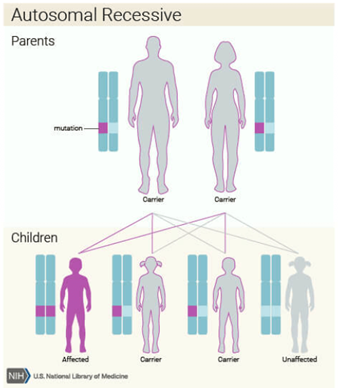
In the case of MPS, an aberrant build-up of specific complex carbohydrates (mucopolysaccharides or glycosaminoglycans) in the arteries, skeleton, eyes, joints, ears, skin, and/or teeth results from a shortage or dysfunction of lysosomal enzymes. Other than that, the build-up also can happen in blood, bone marrow, central nervous system, spleen and in liver.
Those organs that were affected by the accumulation can lead to a very dangerous situation especially for those that have this disorder because it leads to sickness and even death.
Mucopolysaccharidosis type I (MPS I)
This type of disorder is caused by the genetic changes especially in the IDUA (alpha-L-iduronidase) gene which causes the lack of the IDUA enzyme. When the level of IDUA enzyme is reduced, glycosaminoglycans (GAGs) aka large sugar molecule will accumulate in the lysosomes cell and cause different organs and tissues to increase in size.
Interestingly, this type of disorder divided into three separate syndromes from most to least severe such as Hurler syndrome (MPS I-H), Hurler-Scheie syndrome (MPS I-HS) and Scheie syndrome (MP I-S). For this type of condition, the symptoms may begin any time during a person’s life.
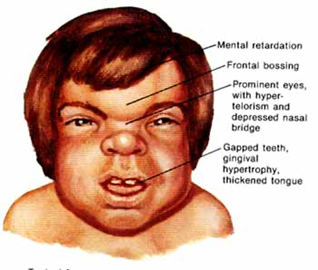
Symptoms include large head, heart valve abnormalities, coarse facial features, hepatosplenomegaly, large tongue, and enlargement of the vocal cords. individuals with severe MPS I, may experience deterioration of intellectual ability, and illness is developing more quickly. Typically, developmental delay is noticeable by age 1, and people who are severely impacted eventually lose fundamental functional skills (developmentally regress).
MPS disorders are irreversible due to this disease being inherited from parents. However, proper treatments can be adopted for patients such as enzyme replacement therapy (ERT), hematopoietic cell transplantation (HCT), and gene therapy. Do consult with the doctors on which treatments are suitable for you.
Witty Charman Medical shall be starting Diagnostic Testing services in newborn children and babies for this disease, using Dried Blood Stains in collaboration with College of American Pathology accredited Laboratories.
Contact us for more details at info@wittycharman.com or at:
- Wendy (+6012-387 8395 / wendy@wittycharman.com)
- Auni (+60 13-233 3646 / auni@wittycharman.com)
- Arissa (+60 12-210 3438 / arissa@wittycharman.com)
Read more on all MPS Types here:
MPS Type II: The Mucopolysaccharidoses Disease Series: Type II – Awam Clinic
MPS Type III: The Mucopolysaccharidoses Disease Series: Type III – Awam Clinic
MPS Type IV: The Mucopolysaccharidoses Disease Series: Type IV – Awam Clinic
References
- Genetic and Rare Diseases Information Center. (2021). Mucopolysaccharidosis type I – About the Disease . National Center for Advancing Translational Sciences. Retrieved September 12, 2022, from https://rarediseases.info.nih.gov/diseases/10335/mucopolysaccharidosis-type-i/
- Clarke, L. A. (2008, January). The mucopolysaccharidoses: a success of molecular medicine. Expert Reviews in Molecular Medicine, 10. https://doi.org/10.1017/s1462399408000550
- MedlinePlus. (2012). Mucopolysaccharidosis type I: MedlinePlus Genetics. MedlinePlus Genetic. Retrieved September 12, 2022, from https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-i/
- NORD – National Organization for Rare Disorders. (2017, June 23). Mucopolysaccharidoses. NORD (National Organization for Rare Disorders). Retrieved September 12, 2022, from https://rarediseases.org/rare-diseases/mucopolysaccharidoses/